- 热门关键词:
- bepaly手机网站
- 缩水甘油
- bepaly意甲
- 巯基乙酸

结构式
| 物竞编号 | 00LZ |
|---|---|
| 分子式 | C17H18FN3O3S |
| 分子量 | 363.41 |
| 标签 | 芦氟沙星, 9-氟-2,3-二氢-10-(4-甲基-1-哌嗪基)-7-氧代-7H-吡啶并[1,2,3-de]-1,4-苯并噻嗪-6-羧, ME-934, ISF-09334, Qari, Monos, Tebraxin, 抗病源性微生物药, 喹诺酮类药物, 药物 |
编号系统
CAS号:101363-10-4
MDL号:暂无
EINECS号:暂无
RTECS号:暂无
BRN号:暂无
PubChem号:暂无
物性数据
暂无
毒理学数据
暂无
生态学数据
暂无
分子结构数据
1、 摩尔折射率:77.70
2、 摩尔体积(cm3/mol):202.8
3、 等张比容(90.2K):599.7
4、 表面张力(dyne/cm):76.4
5、 极化率(10-24cm3):30.80
计算化学数据
1.疏水参数计算参考值(XlogP):无
2.氢键供体数量:1
3.氢键受体数量:8
4.可旋转化学键数量:2
5.互变异构体数量:无
6.拓扑分子极性表面积89.4
7.重原子数量:25
8.表面电荷:0
9.复杂度:608
10.同位素原子数量:0
11.确定原子立构中心数量:0
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
盐酸芦氟沙星(Rufloxacin Hydrochloride):C17H18FN3O3S?HCl。[106017-08-7]。在乙醇-水中结晶,熔点322~324℃。急性毒性LD50兔子,雄大鼠,雌大鼠(mg/kg):660,631,501口服。
贮存方法
暂无
合成方法
方法1:由2,3,4-三氯硝基苯经氟化而得2,4-二氟-3-氯硝基苯,与巯基乙醇缩合后经还原、溴化、环合得苯并噻嗪。苯并噻嗪与EMME缩合后,再经Gould.Jacobs环化反应形成喹诺酮母环,再经水解、氧化、与N-甲基哌嗪缩合、还原而得芦氟沙星。与盐酸成盐可得盐酸芦氟沙星。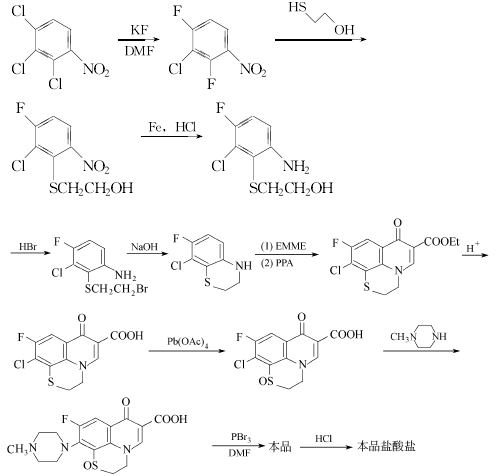
2,4-二氟-3-氯硝基苯也可和巯基乙酸乙酯缩合,再经硝基还原、环合而得苯并噻嗪。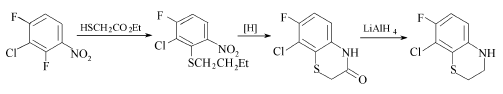
方法2:由2,3,4,5-四氟苯甲酸为起始原料,制得四氟苯甲酰乙酸乙酯后,经与N-甲基哌嗪缩合,再与N,N-二甲基甲酰胺缩二甲醇(DMFA)反应后经置换、分子内亲核取代、环化生成喹诺酮母环,再水解成盐得盐酸芦氟沙星。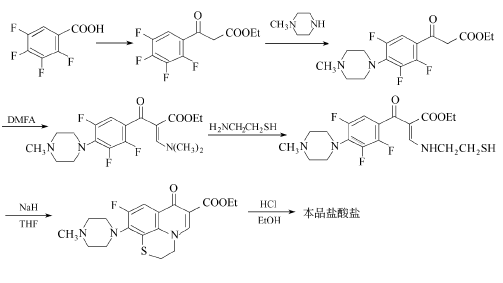
方法3:2,3,4-三氟硝基苯和2-巯基乙醇溶于无水乙醇,0℃以下滴加三乙胺,反应至完全。减压浓缩,加水,用氯仿提取。提取液浓缩,得中间体(Ⅰ),直接用于下步反应。
中间体(I)、乙醇、氯化铵和水混合,在40℃下加入铁粉和稀盐酸,反应至完全。回收乙醇,用乙酸乙酯提取,浓缩后得中间体(II)。
中间体(II)和40%氢溴酸,在120℃下反应至完全。减压蒸出氢溴酸,得中间体(Ⅲ)。
中间体(Ⅲ)和乙醇混合,加热,用碳酸钠调至Ph=9,在50℃下反应。回收乙醇,用氯仿提取,浓缩后得中间体(Ⅳ)。
中间体(Ⅳ)和EMMF,在120℃下反应,加入PPA,升至160℃反应。冷却,加水,再温热。冷却,滤取固体,用二甲基甲酰胺重结晶,得中间体(V),收率69%。中间体(V)和40%氟硼酸混合,在约50℃下反应。冷却,过滤,用二甲基甲酰胺和甲醇(5:2)重结晶,得中间体(VI),收率96%。
中间体(VI)、N-甲基哌嗪、三乙胺和二甲基甲酰胺,在50~60℃下反应。回收溶剂,加入5%氢氧化钠溶液溶解剩余物,过滤。滤液用2mol/L盐酸调至Ph=7。滤取固体,用30%乙醇重结晶,得芦氟沙星,收率76.9%,熔点300~301℃。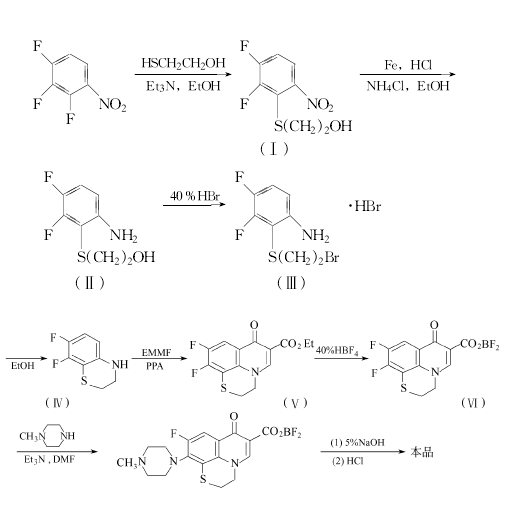
用途
为长效喹诺酮类抗菌药。对草兰阳性和阴性菌,尤其对肠杆菌有显著的抗菌活性。绿脓杆菌,D族链球菌和多数厌氧菌对本品耐药。对细菌性前列腺炎有很高的疗效,耐受性好。用于敏感菌引起的泌尿道感染,下呼吸道感染。
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
None
备注
None
表征图谱

暂无图谱
统计数据
共收录beplay体育首页
数据
147579 条

-
微信扫一扫
关注:物竞beplay体育首页 数据库
-
物竟数据库 手机版
国内首款化工类专业手机应用
-
微博账号
wjhxp
快速导航
关于物竞
Beplay官网网页
是一个全面、专业、专注,并且免费的中文beplay体育首页
信息库,为学生、学者、beplay体育首页
研究机构、检测机构、beplay体育首页
工作者提供专业的beplay体育首页
平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、beplay体育首页
俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们
-
微信账号:物竞beplay体育首页 数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号