- 热门关键词:
- bepaly手机网站
- 缩水甘油
- bepaly意甲
- 巯基乙酸
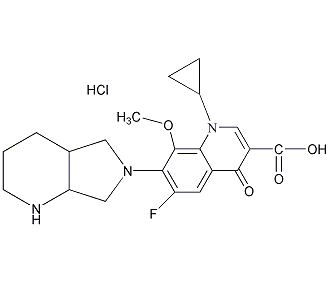
结构式
| 物竞编号 | 00LV |
|---|---|
| 分子式 | C21H24FN3O4 |
| 分子量 | 401.43 |
| 标签 | (4aS-cis)-1-环丙基-6-氟-1,4-二氢-8-甲氧基-7-(六氢-6H-吡咯[3,4-b] 吡啶-6-基)-4-氧-3-喹啉羧酸, Avelox, Actira, Avaiox, Proflox, Octegra, (4αS-cis)-1-Cydopropyl-6-fluoro-1,4-dihydro-8-methoxy-7-(octahydro-6H-pyrrolo[3,4-b]pyridin-6-yl)-4-oxo-3-quinolinecarboxylic acid, 喹诺酮类药物, 抗病源性微生物药 |
编号系统
CAS号:151096-09-2
MDL号:暂无
EINECS号:暂无
RTECS号:暂无
BRN号:暂无
PubChem号:暂无
物性数据
性质:晶体
熔点(ºC):203~208
毒理学数据
暂无
生态学数据
暂无
分子结构数据
1、 摩尔折射率:101.82
2、 摩尔体积(cm3/mol):284.9
3、 等张比容(90.2K):795.0
4、 表面张力(dyne/cm):60.5
5、 极化率(10-24cm3):40.36
计算化学数据
1.疏水参数计算参考值(XlogP):0.6
2.氢键供体数量:2
3.氢键受体数量:8
4.可旋转化学键数量:4
5.互变异构体数量:无
6.拓扑分子极性表面积82.1
7.重原子数量:29
8.表面电荷:0
9.复杂度:727
10.同位素原子数量:0
11.确定原子立构中心数量:2
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:1
性质与稳定性
暂无
贮存方法
暂无
合成方法
推荐如下合成路线,中间体1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸与硼的螯合物按文献[8]方法制备.
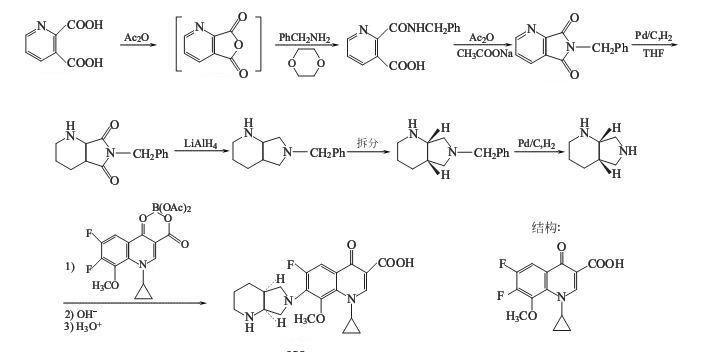
(1).6-苄基-吡咯并[3,4-b]吡啶-5,7-二酮的制备
在反应瓶中加入75.15g(0.45mol)和乙酸酐170ml(1.8mol),加热微回流搅拌反应4.5h.减压浓缩至干,剩余物加二烷100ml,搅拌加热至70 ºC,待溶解后减压蒸除溶剂,得白色固体.直接进行下步反应.
在反应瓶中加入二烷200ml和上步骤所得白色固体,室温搅拌下滴加苄基胺(PhCH2NH2)49.2ml(0.45mol),约30min滴完,继续在室温搅拌1.5h.减压浓缩至干,剩余物中加入乙酸酐191ml(2.03mol)和乙酸钠5.54g(0.068mol),升温至125 ºC
,在该温度保持搅拌3.5h.冷至室温,过滤,滤饼水洗,乙醇洗,抽干,干燥,得白色固体92.3g.滤液减压浓缩至体积40ml,室温放置过夜析晶,回收6.02g,共得98.3g,收率91.8%,mp161~163 ºC.
(2) 6-苄基-六氢-吡咯并[3,4-b]吡啶-5,7-二酮的制备
在高压釜中加入20g(0.084mol)、THF150ml和5%Pd/C 2.5g,用N2置换釜内空气三次,氢气置换N2三次,充氢(85 ºC/8Mpa)反应,直至不吸氢为止(大约5h),冷却,放余压,滤除反应液中Pd/C,滤液减压浓缩至干,得黄色油状物粗品20.42g,不要纯化,直接进行下步反应.
(3)6-苄基-八氢-吡咯并[3,4-b]吡啶的制备
在反应瓶中于氮气保护下加入THF 500ml和LiAlH4 10.3g(0.27mol),并在搅拌下和氮气保护下滴加上步无水THF(60ml)溶液,加毕,搅拌缓慢升温至回流,搅拌回流16h.冷至室温,慢慢加入水10.5ml和THF100ml,滴加5%NaOH25ml,加毕,搅拌回流1h.冷却至室温,过滤,滤渣用THF洗,滤液及洗液合并,减压浓缩至干,得棕色黏稠状液体17.0g,收率93.7%.
(4) (S,S)-6-苄基-八氢-吡咯并[3,4-b]吡啶的制备在
反应瓶中加入L-(+)-酒石酸21.1g(0.141mol)、DMF60ml,搅拌加热到80 ºC溶解,30min内滴加[19.0g(0.088mol)]的DMF(20ml)溶液,同温度下搅拌1h,冷却至室温后再搅拌1h,放置过夜.过滤,滤饼用DMF洗,乙醇洗,用乙二醇单甲醚重结晶,得(R,R)- (S,S)-6-苄基-八氢-吡咯并[3,4-b]吡啶的L-酒石酸盐白色晶体13.6g,收率84.4%,mp112~120 ºC .
合并滤液和重结晶洗液,减压浓缩至干,剩余物中加水50ml,搅拌下加入30%NaOH水溶液50ml,干90~100 ºC 搅拌1h.冷却至室温,除去少量漂浮的黑色黏稠物,用环己烷(150ml*3)提取,有机层用无水硫酸钠干燥,过滤,滤液减压浓缩,得棕色液体10g,待用.
在反应瓶中加入D-(-)-酒石酸11.0g(0.073mol)和DMF 35ml,搅拌加热至80 ºC 溶解,于30min内滴加上述所得之棕色液体的DMF(15ml)溶液,同温搅拌1h.自然冷却至室温后再搅拌1h,放置过夜.过滤,滤饼用(S,S)- (S,S)-6-苄基-八氢-吡咯并[3,4-b]吡啶的D-酒石酸盐12.5g,收率77.6%,mp112~120 ºC
(S,S)- (S,S)-6-苄基-八氢-吡咯并[3,4-b]吡啶的D-酒石酸盐12.5g(0.034mol)加
到反应瓶,加水40ml,搅拌溶解,搅拌下加入30%NaOH水溶液11ml,室温搅拌1h.用环己烷(150ml*3)提取,有机层用无水硫酸钠干燥,过滤,滤液减压浓缩至干,得黄色黏稠液(S,S)-6-苄基-八氢-吡咯并[3,4-b]吡啶 6.6g,收率89.4%.
(5).(S,S)-八氢-6H-吡咯并[3,4-b]吡啶的制备
在高压釜加入(S,S)-6-苄基-八氢-吡咯并[3,4-b]吡啶 6.5g(0.03mol)和甲醇
50ml,搅拌溶解,加入5%Pd/C 1.35g,于90 ºC /9MPa下氢化(操作同(2))16h.过滤,滤液减压浓缩至干(浴温<45 ºC ).剩余物溶于水20ml,用环己烷(20ml*3)提取,有机层用水反提取(30ml*3),合并水层,加入NaOH 2g,用氯仿(30ml*3)提取,有机层用无水硫酸钠干燥,过滤,滤液减压浓缩至干,得浅黄色黏液.3.4g,收率89.9%.
(6). (1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸-O3,O4)双(乙酰氧)-硼酸盐(或硼螯合物)的制备
在反应瓶中加入硼酸57.2g(0.925mol)和氯化锌1.24g,再向该混合物中加入乙酸酐300ml,搅拌升温至110 ºC ,在该温度搅拌1.5h.再向该反应混合物中加入冰醋酸400ml,在同温度下搅拌1h.放冷后,在50~60 ºC加入1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸 200g(0.619mol),随后加入200ml冰醋酸,在同温度下搅拌5h.将反应混合物减压浓缩,剩余油状物中加入8L冰水,产生沉淀,过滤收集,再悬浮至3L水中,过滤,干燥,得到(1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸-O3,O4)双(乙酰氧)-硼酸盐(或硼螯合物) 249g,它是一亮橙黄色粉末状物,mp113~117 ºC
(7) 莫西沙星的合成
在反应瓶中加入(1-环丙基-6,7-二氟-1,4-二氢-8-甲氧基-4-氧代-3-喹啉羧酸-O3,O4)双(乙酰氧)-硼酸盐(或硼螯合物) 2.0g(4.73mmol)和乙腈10ml,搅拌溶解,再加入三乙胺0.5ml和(S,S)-八氢-6H-吡咯并[3,4-b]吡啶 0.66g(5.24mmol),搅拌回流3h.减压浓缩至干,剩余物加石油醚,捣碎分散固化,得黄色固体过滤,滤饼用石油醚洗,抽干,溶于7%NaOH溶液13.5ml,于80 ºC 搅拌3h,冷至室温,过滤,滤液用5%乙酸水溶液调至PH7,过滤,滤饼用5%乙酸水溶液溶解,过滤,滤液用5%NaOH水溶液调至PH7,过滤,滤饼用水洗,乙醇洗,抽干,干燥,得淡黄色固体莫西沙星 1.53g,收率80.5%,mp203~207 ºC
用途
1.氟喹诺酮类抗菌药。DNA拓扑异构酶抑制剂,可用于治疗金葡菌、流感杆菌、肺炎球菌等引起的社会获得性肺炎,慢性支气管炎急性发作,急性窦炎等。
2.属第四代喹诺酮类抗菌药物,是新一代抗菌谱广的抗生素.该品对常见的呼吸道病菌,如肺炎链球菌、嗜血流感杆菌、卡他莫拉汉菌以及部分金黄色葡萄球菌都具有很强的抗菌活性.特别是对肺炎链球菌,抗菌作用强大.临床用于治疗急性窦腺炎、慢性支气管炎的急性发作)社区获得性肺炎,以及无并发症的皮肤感染和皮肤软组织感染.本品特点是几乎没有光敏反应,具有良好的组织穿透力,在肺组织中也可达到很高浓度,是治疗呼吸道感染较好的药物.
安全信息
危险运输编码:暂无
危险品标志: 有害
有害
安全标识:S36/S37
文献
[1] Merck Index.13 th:6316 [2] Woodcock J M,et al.Antimicrob Ag Chemother, 1997,41:101 [3]Ji B,et al, Antimicrob Ag Chemother,1998,42:2066 [4]刘九雨等。国外医药—抗生素分册,2002,23:274-278. [5]EP,550903.1993 [6] EP,603887.1994 [7] EP,241206.1987. [8]EP,464823.1992. [9]刘明亮等。中国医药工业杂志,2004,35:129-131. [10] 四川美康医药软件研究开发有限公司编著。药物临床信息参考。成都:四川出版集团四川科学技术出版社,2006.235-236
备注
暂无
表征图谱

暂无图谱
统计数据
共收录beplay体育首页
数据
147579 条

-
微信扫一扫
关注:物竞beplay体育首页 数据库
-
物竟数据库 手机版
国内首款化工类专业手机应用
-
微博账号
wjhxp
快速导航
关于物竞
Beplay官网网页
是一个全面、专业、专注,并且免费的中文beplay体育首页
信息库,为学生、学者、beplay体育首页
研究机构、检测机构、beplay体育首页
工作者提供专业的beplay体育首页
平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、beplay体育首页
俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们
-
微信账号:物竞beplay体育首页 数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号