- 热门关键词:
- bepaly手机网站
- 缩水甘油
- bepaly意甲
- 巯基乙酸
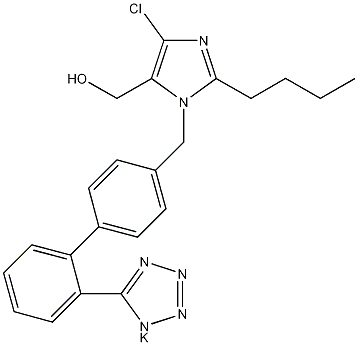
结构式
| 物竞编号 | 0YBB |
|---|---|
| 分子式 | C22H23ClN6O |
| 分子量 | 422.91 |
| 标签 | 2丁基-4-氯-1-([2'-(1H-四唑-5)(1,1''-联苯)-4-基]甲基)-1H-咪唑-5-甲醇单钾盐, 2-Butyl-4-chloro-1-{[2'-(1H-tetrazol-5-yl)(1,1'-biphenyl)-4-yl]methyl}-1H-imidazole-5-methanol monopotassium salt, 拮抗剂 |
编号系统
CAS号:124750-99-8
MDL号:MFCD02092704
EINECS号:暂无
RTECS号:NI6755100
BRN号:5845770
PubChem号:暂无
物性数据
1. 性状:亮黄色固体。
2. 密度(g/mL,20℃):未确定
3. 相对蒸汽密度(g/mL,空气=1):未确定
4. 熔点(ºC):183.5~184.5
5. 沸点(ºC,常压):未确定
6. 沸点(ºC,KPa):未确定
7. 折射率:未确定
8. 闪点(ºC):未确定
9. 比旋光度(º):未确定
10. 自燃点或引燃温度(ºC):未确定
11. 蒸气压(Pa,20ºC):未确定
12. 饱和蒸气压(KPa,20ºC):未确定
13. 燃烧热(KJ/mol):未确定
14. 临界温度(ºC):未确定
15. 临界压力(KPa):未确定
16. 油水(辛醇/水)分配系数的对数值:未确定
17. 爆炸上限(%,V/V):未确定
18. 爆炸下限(%,V/V):未确定
19. 溶解性:未确定
毒理学数据
生态学数据
暂无
分子结构数据
1、摩尔折射率:无可用的
2、摩尔体积(cm3/mol):无可用的
3、等张比容(90.2K):无可用的
4、表面张力(dyne/cm):无可用的
5、介电常数:无可用的
6、极化率(10-24cm3):无可用的
7、单一同位素质量:460.118069 Da
8、标称质量:460 Da
9、平均质量:461.001 Da
计算化学数据
1、 疏水参数计算参考值(XlogP):
2、 氢键供体数量:1
3、 氢键受体数量:6
4、 可旋转化学键数量:8
5、 互变异构体数量:2
6、 拓扑分子极性表面积(TPSA):77.7
7、 重原子数量:31
8、 表面电荷:0
9、 复杂度:526
10、同位素原子数量:0
11、确定原子立构中心数量:0
12、不确定原子立构中心数量:0
13、确定化学键立构中心数量:0
14、不确定化学键立构中心数量:0
15、共价键单元数量:2
性质与稳定性
暂无
贮存方法
暂无
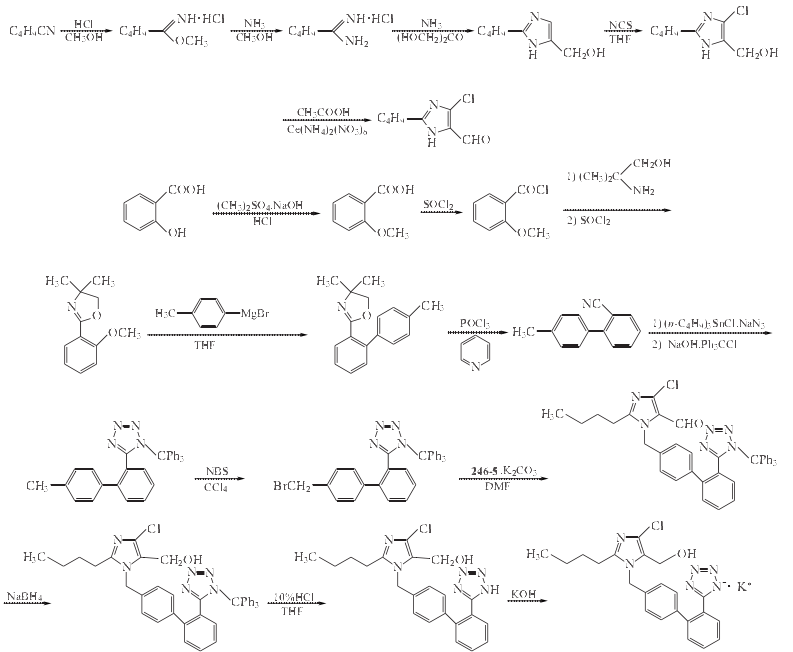
合成方法
1. 戊脒盐酸盐的制备
在反应瓶中加入戊腈30g(0.361mol)和无水甲醇12g(0.375mol),搅拌溶解,在冰盐浴冷却下,通入干燥的HCl气体至增加质量18g,加盖瓶塞,侧瓶颈接无水CaCl2干燥管,放置5天,得白色结晶亚氨酯.将亚氨酯用无水甲醇溶解,加入用氨饱和的无水甲醇溶液(NH4浓度不低于9%),室温搅拌6h.抽滤,滤饼为生成的NH4Cl,弃去,滤液蒸除溶剂后得戊脒盐酸盐15g,收率30.4%(从戊腈计两步收率).该品冷却时结晶#常温时融化.戊脒盐酸盐冷却时结晶成苦味酸盐,在乙酸乙酯中重结晶#得黄色针状结晶,mp192~193ºC.
2. 2-丁基-5-羟甲基咪唑的制备
在反应瓶中加入戊脒盐酸盐5.2g(38mmol)和无水甲醇25ml,搅拌下通入NH3,使其反应液增重6g,加入二羟丙酮3.8g(42mmol)的无水甲醇20ml溶液.将其加进高压釜,室温搅拌1h.升温至40ºC,在该温度搅拌45min,升温至70ºC,在70ºC搅拌反应6h.冷却,减压蒸除溶剂,剩余物中加入适量乙腈,加热至沸,然后倾倒出乙腈溶液,重复操作几次,将残留的氯化铵弃去,合并乙腈溶液,浓缩至干,剩余物用乙酸乙酯溶解后,用硅胶柱色谱纯化,乙酸乙酯洗脱,目的物组分流出液经浓缩后,得黄色油状物,用乙酸乙酯/石油醚(bp60~90ºC)结晶,得2-丁基-5-羟甲基咪唑3.8g,为浅黄色固体,mp86~88 ºC,收率70%.
3. 2-丁基-4-氯-5-羟甲基咪唑的制备
在反应瓶中加入2-丁基-5-羟甲基咪唑1g(7mmol)、无水THF10ml,搅拌溶解#再加入N-氯代丁二酰亚胺(NCS)1g(7mmol),在40 ºC搅拌反应1h.蒸除溶剂#加入乙酸乙酯和水混合液(1:1,质量比),充分振摇,静置分层,分出乙酸乙酯层,无水NaSO4干燥,过滤,滤液减压蒸除溶剂,剩余物用石油醚(bp60~90)研磨,得白色固体2-丁基-4-氯-5-羟甲基咪唑0.7g,收率57%,mp138~140 ºC.
4. 2-丁基-4-氯-5-甲酰基咪唑的制备
在反应瓶中加入2-丁基-4-氯-5-羟甲基咪唑50g(265mmol)和冰乙酸150ml,搅拌溶解,于25º搅拌滴加1mol/L硝酸铈铵水溶液595ml(595mmol),滴加时控制反应液温度25~30ºC,滴毕,在25ºC搅拌反应3h.将反应混合物冷至0ºC,加入50%NaOH水溶液210ml,产生沉淀,调至Ph6,静置后过滤,干燥得2-丁基-4-氯-5-甲酰基咪唑38.1g,收率77%,mp92.5~93.5ºC.
5. 4,4-二甲基-2-(2-甲氧基苯基)恶唑啉的制备
在反应瓶中加入2-甲氧基苯甲酸(按文献[8~10]的方法制备)30g(0.20mol)和氯化亚砜50ml(81.9g,0.69mol),在20ºC搅拌反应18h.在真空下蒸除过量的氯化亚砜,粗品2-甲氧基苯甲酰氯,其真空蒸馏,集81~83ºC/93.33Pa馏分,2-甲氧基苯甲酰氯32g,收率95%,为无色液体,于下步反应.
在反应瓶中加入2-氨基-2-甲基-1-丙醇20g(0.224mol)和二氯甲烷100ml,搅拌下冷至0 ºC,滴加2-甲氧基苯甲酰氯17g(0.100mol)溶于二氯甲烷50ml的溶液,滴毕,升至20ºC搅拌反应2h.减压蒸除溶剂,剩余物加水稀释,析出固体,过滤,滤饼用水洗,干燥,得白色固体20.5g,该产品为粗制酰胺中间体.
在反应瓶加入上述酰胺中间体20.5g(0.092mol)和氯化亚砜22ml(36g,0.302mol),于25ºC搅拌反应1h.将反应液倒入乙醚中,析出的固体过滤收集,用乙醚洗涤,干燥.然后将其溶于水中,用1mol/LNaOH水溶液调至pH10, 用乙醚提取数次,合并有机相,无水Na2SO4干燥,过滤,滤液真空下浓缩,得4,4-二甲基-2-(2-甲氧基苯基)恶唑啉18.0g,收率88%[从2-甲氧基苯甲酰氯计]mp70~72ºC].
6. 4,4-二甲基-2-(4’-甲基联苯-2-基)恶唑啉的制备
在无水干燥反应瓶中加入无水THF200ml和镁屑2.50g(0.103mol),在N2保护下,于20ºC搅拌下滴加4-溴甲苯13ml(0.106mol) ,滴毕,搅拌反应直至镁屑全部溶解(约1h),得对甲苯基溴化镁溶液.
在反应瓶中加入4,4-二甲基-2-(2-甲氧基苯基)恶唑啉10.0g(48.7mmol)和无水THF100ml,搅拌溶解,于20ºC滴加上述对甲苯基溴化镁溶液,加毕,在20ºC搅拌反应2h.将反应混合物倒入饱和的NH4Cl水溶液中,形成的乳浊液用乙酸乙酯提取,有机相用无水Na2SO4干燥,过滤,滤液减压浓缩,得油状物粗品,经硅胶柱色谱纯化,用35%的乙酸乙酯/己烷洗脱,经后处理得4,4-二甲基-2-(4’-甲基联苯-2-基)恶唑啉11.8g,收率91%,为无色液体.
7. 2-氰基-4’-甲基联苯的制备
在反应瓶中加入4,4-二甲基-2-(4’-甲基联苯-2-基)恶唑啉243.2g(0.917mol)和吡啶975ml,于10ºC搅拌下滴入POCl3 172ml(1.84mol),在滴加过程中控制内温不超过15ºC.滴毕,升温至100ºC,在该温度搅拌反应3h.冷至室温,加水终止反应,得到的乳浊液用乙酸乙酯提取,有机相合并,用水洗,用10%CuSO4水溶液洗和用饱和食盐水洗,无水MgSO4干燥,过滤,滤液减压浓缩,剩余物用庚烷重结晶,得无色针状结晶2-氰基-4’-甲基联苯169.6g,收率96%,mp48.0~49.5ºC.
8. N-(三苯基甲基)-5-(4’-甲基联苯-2-基)-四氮唑的制备
在反应瓶中加入2-氰基-4’-甲基联苯9.0g(46.6mmol)、三丁基氯化锡16.4g(50.4mmol)、叠氮化钠30g(46.2mmol)和甲苯35ml,搅拌回流70h.加入甲苯30ml稀释,并冷至20ºC.再加入10mol/LNaOH水溶液5.5ml(55mmol)和三苯基氯甲烷13.5g(48.4mmol),在20ºC搅拌反应3h.向反应液加入蒸馏水35ml和庚烷70ml,冷至0ºC搅拌1.5h.将浆状物过滤,固体用水洗和庚烷/甲苯(1:1)洗涤,抽干,得粗制品N-(三苯基甲基)-5-(4’-甲基联苯-2-基)-四氮唑,将其溶于二氯甲烷,然后依次用0.4mol/L NaOH水溶液、水和饱和食盐水洗涤,无水Na2SO4干燥,过滤,滤液减压浓缩,得N-(三苯基甲基)-5-(4’-甲基联苯-2-基)-四氮唑15.1g,收率68%,mp163~166ºC.
9. N-(三苯基甲基)-5-[4’-(溴甲基)联苯-2-基]-四氮唑的制备
在反应瓶中加入N-(三苯基甲基)-5-(4’-甲基联苯-2-基)-四氮唑31.0g(65mmol)、N-溴代丁二酰亚胺(NBS)11.50g(65mmol)和四氯化碳390ml,搅拌回流3h.冷至40ºC,过滤,滤液真空浓缩回收溶剂,得到N-(三苯基甲基)-5-[4’-(溴甲基)联苯-2-基]-四氮唑粗品,粗品不需进一步纯化,可用于下步反应.将粗品加入二异丙醚捣碎研磨,得到N-(三苯基甲基)-5-[4’-(溴甲基)联苯-2-基]-四氮唑纯品33.10g,收率92%,为类白色固体,mp135~138 ºC.
10. 2-丁基-4-氯-5-甲酰基-1-[[[2’-(三苯甲基)四氮唑-5-基]联苯-4-基]甲基]咪唑的制备
在反应瓶中加入2-丁基-4-氯-5-甲酰基咪唑3.27g(17.5mmol)、N-(三苯基甲基)-5-[4’-(溴甲基)联苯-2-基]-四氮唑10.73g(19.3mmol)、K2CO3 4.83g(35mmol)和DMF100ml,在25 ºC搅拌反应24h.过滤,滤液减压浓缩,
剩余物加水稀释,用乙酸乙酯提取,合并有机相,用水洗)饱和盐水洗,无水MgSO4干燥,过滤,滤液浓缩,浓缩液经硅胶柱色谱纯化,用乙酸乙酯/甲苯洗脱,经后处理得2-丁基-4-氯-5-甲酰基-1-[[[2’-(三苯甲基)四氮唑-5-基]联苯-4-基]甲基]咪唑5.69g,收率49%.
11. 2-丁基-4-氯-5-(羟甲基)-1-[[2’-(三苯甲基)四氮唑-5-基]联苯-4-基]甲基]咪唑的制备
在反应瓶中加入2-丁基-4-氯-5-甲酰基咪唑3.57g(21mmol)、四丁基溴化磷0.71g(21mmol)、10mol/LNaOH水溶液4.2ml(42mmol)、水15ml和二氯甲烷70ml,搅拌下加入N-(三苯基甲基)-5-[4’-(溴甲基)联苯-2-基]-四氮唑11.65g(21mmol)溶于二氯甲烷100ml的溶液,于25ºC搅拌反应24h.向反应混合物加入硼氢化钠(NaBH4)0.79g(21mmol),再继续搅拌反应24h.加水200ml,静置分层,分出有机相,用水(200ml×2)洗,无水MgSO4干燥,过滤,滤液减压蒸除溶剂,得黄色玻璃状固体2-丁基-4-氯-5-(羟甲基)-1-[[2’-(三苯甲基)四氮唑-5-基]联苯-4-基]甲基]咪唑13.3g,用硝基甲烷重结晶,得2-丁基-4-氯-5-(羟甲基)-1-[[2’-(三苯甲基)四氮唑-5-基]联苯-4-基]甲基]咪唑精品类白色粉末7.52g,收率54%,mp167~169ºC.
12. 氯沙坦钾的合成
在反应瓶中加入化合物2-丁基-4-氯-5-(羟甲基)-1-[[2’-(三苯甲基)四氮唑-5-基]联苯-4-基]甲基]咪唑9.20g(13.8mmol)、10%盐酸100ml和THF200ml,于25ºC搅拌反应4h.加入过量的10%NaOH水溶液(约100ml)减压蒸除溶剂,剩余物用水溶解,过滤除去副产物三苯甲醇,滤液用盐酸调至Ph3,析出沉淀,过滤收集沉淀,得氯沙坦粗制品,重结晶后得氯沙坦氯沙坦钾5.18g,收率89%,mp183.5~184.5ºC.
用途
本品是第一个强力)选择性的非肽类血管紧张素Ⅱ(ATⅡ)、AT1受体拮抗剂.为口服抗高血压药.临床应用于中度、轻度高血压患者,口服氯沙坦钾50~100mg/天,降压效果显著;重度高血压患者,需与利尿药物或其他抗高血压药物合用.应用本品没有ACE抑制剂产生的咳嗽副作用.与钙通道阻滞剂不同,本品不引起踝关节水肿、头痛和心动过速.另有报道,本品用于心衰的治疗也在临床研究中.
安全信息
危险运输编码:暂无
危险品标志:暂无
安全标识:暂无
危险标识:暂无
文献
【1】 Merck Index.13th:5604. 【2】 罗明生主编.现代临床药物大典.成都:四川科学技术出版社,2001:522. 【3】 US,4355040.1982. 【4】 EP,043159Az.1990. 【5】 Dziuron P,et al.Arch Pharm,1974,307:470. 【6】 US,4340598.1982. 【7】 Carni D J,et al.J Med Chem,1991,34:2525-2547. 【8】 Engley M.Annalender Chemine,1915,408:212. 【9】 Dippy J F J,et al.J Chem Soc,1937:1426. 【10】 Dippy I M,Terahedrom Lett,1981,22:1013. 【11】 韩长日等.中国医药工业杂志,1991,22:320. 【12】 Meyers A I,et al.J Am Chem Soc,1975,97:7383. 【13】 EP,253310.1988. 【14】 US,5138069.1992. 【15】 周伟澄主编.高等药物化学选论.北京:化学工业出版社,2006:299. 【16】 MchIntyre M,et al.Pharmacol Ther,1997,74:181-194. 【17】 Wong P C,et al.Am J Hypertens,1991,4:271s-353s.
备注
暂无
表征图谱

暂无图谱
统计数据
共收录beplay体育首页
数据
147579 条

-
微信扫一扫
关注:物竞beplay体育首页 数据库
-
物竟数据库 手机版
国内首款化工类专业手机应用
-
微博账号
wjhxp
快速导航
关于物竞
Beplay官网网页
是一个全面、专业、专注,并且免费的中文beplay体育首页
信息库,为学生、学者、beplay体育首页
研究机构、检测机构、beplay体育首页
工作者提供专业的beplay体育首页
平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、beplay体育首页
俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们
-
微信账号:物竞beplay体育首页 数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号