- 热门关键词:
- bepaly手机网站
- 缩水甘油
- bepaly意甲
- 巯基乙酸
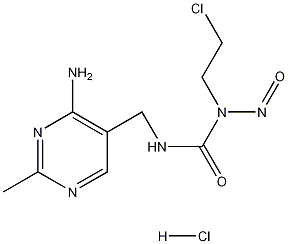
结构式
| 物竞编号 | 0TW6 |
|---|---|
| 分子式 | C9H14Cl2N6O2 |
| 分子量 | 309.15 |
| 标签 | 1-[(4-氨基-2-甲基-5-嘧啶基)甲基]-3-(2-氯乙基)-3-亚硝基脲盐酸盐, 里莫斯定, 嘧啶亚硝脲, 1-(4-Amino-2-methyl-5-pyrimidinyl)methyl-3-(2-chloroethyl)-3-nitrosoureahydrochloride, 抗肿瘤药物 |
编号系统
CAS号:55661-38-6
MDL号:暂无
EINECS号:暂无
RTECS号:YR8450000
BRN号:暂无
PubChem号:暂无
物性数据
1.性状:白至亮黄色结晶性粉末, 无臭。
2.溶解性:在湿空气几乎不溶于氯仿、乙醚、乙酸乙酯、苯、正己烷。
毒理学数据
LD50(mg/kg):小鼠,静脉注射62;大鼠,静脉注射46
生态学数据
暂无
分子结构数据
暂无
计算化学数据
1.疏水参数计算参考值(XlogP):无
2.氢键供体数量:3
3.氢键受体数量:6
4.可旋转化学键数量:4
5.互变异构体数量:6
6.拓扑分子极性表面积114
7.重原子数量:19
8.表面电荷:0
9.复杂度:292
10.同位素原子数量:0
11.确定原子立构中心数量:0
12.不确定原子立构中心数量:0
13.确定化学键立构中心数量:0
14.不确定化学键立构中心数量:0
15.共价键单元数量:2
性质与稳定性
暂无
贮存方法
暂无
合成方法
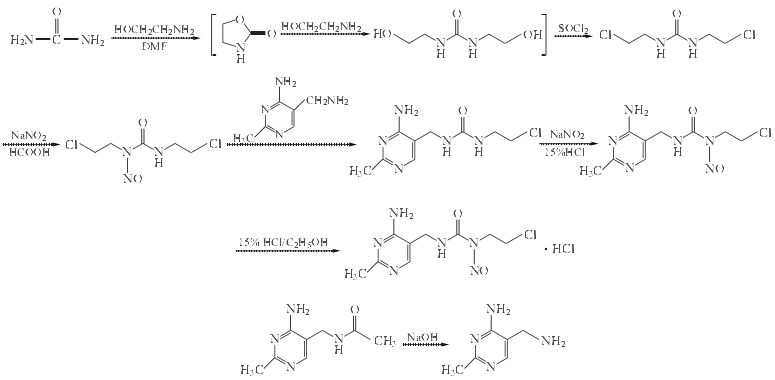
1. 1,3-双(2-氯乙基)脲的制备
在反应瓶中加入尿素90g(1.5mol)、2-氨基乙醇92g(1.5mol) 和DMF750ml, 搅拌加热至回流, 搅拌回流反应8~9h. 减压浓缩回收DMF, 剩余物再加入2-氨基乙醇66g(1.1mol), 搅拌回流2h. 冷至0 ºC, 搅拌下滴加氯化亚砜195ml(319.4g,2.68mol)滴毕, 放置过夜. 将反应混合物再加热至100~120 ºC, 搅拌反应2~3h, 析出固体, 将结块物料小心捣碎, 过滤, 水洗滤饼, 抽干, 粗品用甲醇重结晶, 得白色固体1,3-双(2-氯乙基)脲86g, 三步收率共32%左右,mp120~3 ºC.
2. 1,3-双(2-氯乙基)-1-亚硝基脲的制备
在反应瓶中加入1,3-双(2-氯乙基)脲86g(0.465mol) 和88%甲酸620ml,搅拌混合并在冰水浴冷却下,滴加亚硝酸钠[74g(1.07mol)]的水(680ml)溶液,于4~4.5h内滴加完.滴毕,继续搅拌反应4~5h,用TLC跟踪,确认1,3-双(2-氯乙基)脲消失,反应到终点,停止反应,置于冰箱过夜.过滤,滤饼用冰水洗涤多次,抽干,真空干燥, 得淡黄色固体1,3-双(2-氯乙基)-1-亚硝基脲77g,收率78%左右,mp30~31 ºC.下步用1,3-双(2-氯乙基)-1-亚硝基脲可不干燥,直接用潮品投料.
3. 1-(4-氨基-2-甲基嘧啶-5-基)-甲基-3-(2-氯乙基)脲的制备
在反应瓶中加入乙酰嘧啶(外购品)49.3g(纯度为91.2%,计纯品45g)(.25mol)和13%NaOH水溶液210ml,加热搅拌至回流温度,搅拌回3.5~4h.TLC跟踪乙酰嘧啶消失,则乙酰嘧啶已转化为2-甲基-5-氨基甲基嘧啶. 冷却至室温,用浓盐酸调至pH10左右,加入上步产物1,3-双(2-氯乙基)-1-亚硝基脲潮湿品(测水分后折算干品50g)(0.234mol),搅拌加热至50~55 ºC,在该温度下搅拌反应4~5h.TLC跟踪,确认1,3-双(2-氯乙基)-1-亚硝基脲消失反应达终点, 冷却至室温, 用13%左右NaOH水溶液调至Ph7~8,静置于冰箱过夜.过滤,饼用水洗数次,抽干,干燥,得淡黄色固体1-(4-氨基-2-甲基嘧啶-5-基)-甲基-3-(2-氯乙基)脲30~32g,收率54%左右,mp165~167 ºC.纯度>95%(HPLC归一法).
4. 尼莫司汀的制备
在反应瓶中加入1-(4-氨基-2-甲基嘧啶-5-基)-甲基-3-(2-氯乙基)脲29g(0.12mol)、15%盐酸280ml,搅拌混合溶解,于冰水浴冷却下加入NaNO210g(0.15mol),分批小量加入,加时控制好内温≤0 ºC,加毕,继续在同温度下搅拌反应3~4h,TLC跟踪, 1-(4-氨基-2-甲基嘧啶-5-基)-甲基-3-(2-氯乙基)脲斑点消失反应达终点,终止反应,如果有大量固体产生,加适量水至其溶解,如仍有极少量不溶物时不再加水,加活性炭2~3g,搅拌30mon. 过滤,滤饼用水洗,滤液用20%~25%的NaCO3水溶液中和至pH到8,析出淡黄色固体,过滤,滤饼用水洗涤数次,抽干,干燥,得淡黄色固体尼莫司汀22g左右,收率约68.5%~70.0%,mp124~125 ºC(分解).
5. 盐酸尼莫司汀的合成
在反应瓶中加入尼莫司汀24.8g(0.08mol)、无水乙醇225ml,搅拌悬浮,加入HCl无水乙醇溶液,加量以使悬浮液至澄清透明为准.加毕继续搅拌反应至析出淡黄色固体为止,反应毕,静置于冰箱2~3h.析出大量固体,过滤,得粗品盐酸尼莫司汀18.5g,收率为75%.
取粗品盐酸尼莫司汀15g,加丙酮225ml,搅拌加热回流,加少量稀盐酸至pH呈酸性,溶液变澄清,加入针用活性炭10~15g,轻微加热回流15~20min.趁热抽滤,滤液经微孔滤膜过滤,抽干,除菌,密封,冰箱静置6h. 析出晶体, 抽滤, 真空(70~ 80。以下)速干燥,得无菌粉10.5g,收率70%(精制收率),mp170~171 ºC (分解).
用途
本品为水溶性亚硝脲类抗肿瘤药物#具有烷化作用,能使细胞内DNA烷基化,引起DNA降解分裂及DNA合成障碍,使细胞的分裂增殖受到抑制或引起细胞死亡.因此分裂增殖快的肿瘤细胞首先受抑制,表现为治疗作用.本品与同类烷化剂相比,毒性小,水溶性好,可供静脉给药,给药后能透过血脑屏障,脑内移行良好,适用于颅内肿瘤治疗.临床用于缓解脑肿瘤、消化道肿瘤(胃癌、肝癌、结、直肠癌)、肺癌、恶性淋巴瘤及慢性白血病的主观或客观症状.
安全信息
危险运输编码:UN 2811
危险品标志: 有毒
有毒
安全标识:S45 S36/S37/S39
危险标识:R25
文献
【1】 Merck Index.13th:6581. 【2】 Saijo N,et al,Cancer Chemother Phamacol,1980,4:165. 【3】 中尾英雄等.药学雒誌,1974,94:1032-1037.(日文). 【4】 罗明生主编.现代临床药物大典.成都:四川科学技术出版社, 【5】 陈芬儿主编.有机药物合成法:第一卷.北京:中国医药科技出版社,1999:866-869. 【6】 DE,2257360.1973. 【7】 US,4003901.1977. 【8】 药学雒誌,1974,94:1932. (日文). 【9】 鹤岛正明等, 药学雒誌,1973,93:1274-1284(日文). 【10】 Johnston T P,et al,J Med Chem,1996,9:892-911. 【11】 Johnston T P,et al,J Med Chem,1963,6:669-681. 【12】 Jkuya Shibata et al.Bull Chem Soc Jpn,1989,62:853-859. 【13】 George W,et al,J Am Chem Soc,1957,79:6180-6183. 【14】 Yoshio Iwakura,et al,J Org Chem,1960,25:1118-1123. 【15】 张荣久等.中国药物化学杂志,1996,6:207-208. 【16】 Gesellschaft Deutscher Chemiker,Beilsteins Handbuch der Organischen Chemie,Berlin,EV,25/12,V134-V136(1993). 【17】 章思规主编.精细有机beplay体育首页 技术手册:下卷.北京:科学出版社,1993:1429. 【18】 王世玉,王普善编著.合成药物与中间体手册.北京:化学工业出版社,2004:553-554.
备注
暂无
表征图谱

暂无图谱
统计数据
共收录beplay体育首页
数据
147579 条

-
微信扫一扫
关注:物竞beplay体育首页 数据库
-
物竟数据库 手机版
国内首款化工类专业手机应用
-
微博账号
wjhxp
快速导航
关于物竞
Beplay官网网页
是一个全面、专业、专注,并且免费的中文beplay体育首页
信息库,为学生、学者、beplay体育首页
研究机构、检测机构、beplay体育首页
工作者提供专业的beplay体育首页
平台进行交流。
数据库采用全中文化服务,完全突破了中英文在化学物质命名、beplay体育首页
俗名、学名等方面的差异,所提供的数据全部中文化,更方便国内从事化学、化工、材料、生物、环境等化学相关行业的工作人员查询使用。
关注我们
-
微信账号:物竞beplay体育首页 数据库

-
微博账号:wjhxp
联系我们
上海市延长路149号上海大学科技园412室
公司总机: 021-56389801
订购电话: 4007001514
传真电话: 021-56389802
客服电话: 021-56332350
电子邮件: wingch@basechem.org
 沪公网安备 31010602001115号
沪公网安备 31010602001115号